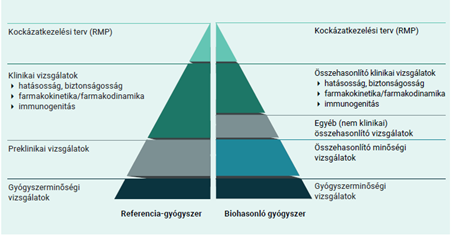
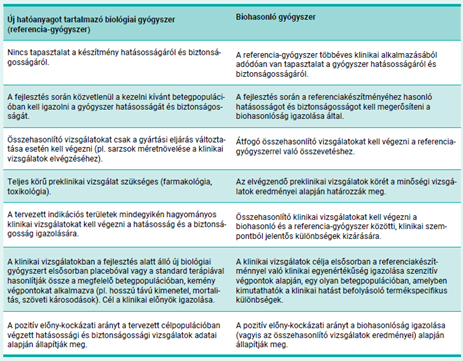
A biohasonló és a biológiai referenciagyógyszert általában ugyanolyan gyógyszerformában és adagban alkalmazzák ugyanazon betegségek kezelésére. Mivel a biohasonló és a biológiai referenciagyógyszer csak hasonló, de teljes azonosságuk a molekula összetettsége miatt nem igazolható, ezért a biohasonlósági alapon történő gyógyszerengedélyezés szigorú tudományos megalapozottságot és nagy körültekintést igényel.
A különféle krónikus megbetegedések gyógyítására szolgáló gyógyszerkészítmények egyre nagyobb hányadát teszik ki a biotechnológiai eredetű gyógyszerek. A nemzetközi előrejelzések azt mutatják, hogy a biotechnológiai eredetű készítmények piaca éves szinten eléri a 28%-ot a teljes, globális gyógyszerfelhasználást tekintve (2020).
A biotechnológiai eredetű készítmények árszintje általában magasabb, mint a hagyományos kémiai úton előállított kismolekulás gyógyszereké, tekintettel a nagyobb kutatás-fejlesztési ráfordításokra, az általuk megcélzott kisebb létszámú betegpopulációkra, a molekuláris szintű komplexitás miatti fokozott biztonságossági követelményekre, valamint ezzel összefüggésben a magasabb előállítási költségekre.
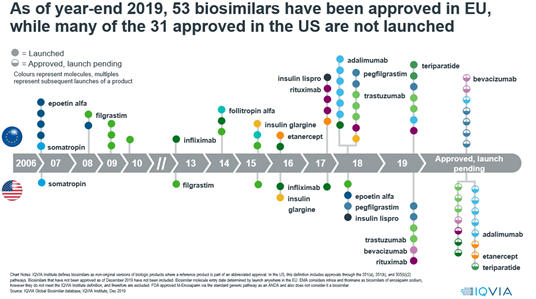
Az első biotechnológiai eredetű készítmények piaci kizárólagossága a 2005 és 2010 közötti időszakban járt le mind az Egyesült Államokban, mind Európában. Az originátor biotechnológiai eredetű készítmény piaci kizárólagosságvesztése után léphetnek piacra a biohasonló (biosimilar) készítmények.
A biohasonló készítmények törzskönyvezése terén az Európai Unió gyakorlata sokkal megelőzi az Egyesült Államokét: az Európai Gyógyszerügynökség (EMA) az EU vonatkozó irányelveit követve, szigorú szabályok mentén, kötelező centralizált eljárásban, folyamatosan végzi ezen készítmények törzskönyvezését, szemben az amerikai Gyógyszer- és Élelmiszerügyi Hivatallal (FDA), amelynek ehhez egyelőre hiányzik a törvényi felhatalmazása.
- Biológiai (referencia) készítmény
- Bioekvivalencia
- Biohasonlóság
- Variabilitás
- Felcserélhetőség
- Immunogenitás
- Extrapolálás
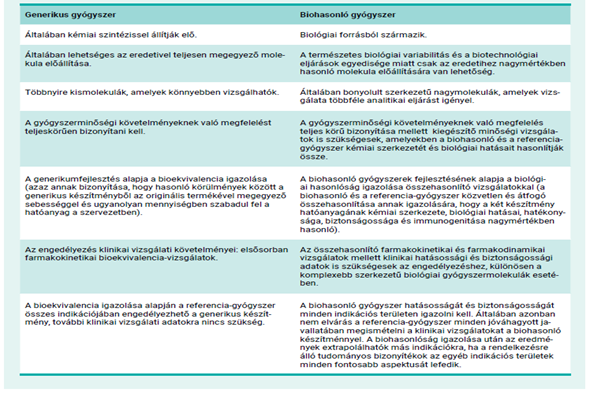
- Generikus vs biohasonló gyógyszer
Az EU-ban már engedélyezett biológiai gyógyszer, amelyet a biohasonló készítményt fejlesztő cég referenciaként választ a minőségi, a biztonságossági és a hatásossági jellemzők közvetlen összehasonlító vizsgálataihoz.
Bioekvivalencia akkor állapítható meg, ha két gyógyszerből hasonló körülmények között ugyanaz a hatóanyag, ugyanolyan sebességgel és ugyanolyan mennyiségben szabadul fel a szervezetben.
A referenciakészítménnyel való nagyfokú hasonlóság igazolása a kémiai szerkezet, a biológiai hatás és a hatékonyság, a biztonságosság és az immunogén potenciál tekintetében.
A biohasonlóság igazolása részletes összehasonlító vizsgálatokkal történik.
Mivel a biohasonló gyógyszereket élő organizmusok felhasználásával állítják elő, kismértékben eltérhetnek a referenciakészítménytől. Ezek a kisebb eltérések azonban klinikai szempontból nem jelentősek, vagyis sem a biztonságosság, sem a hatásosság terén nem várhatók eltérések az alkalmazásuk során. A természetes variabilitás a biológiai gyógyszerek jellemző sajátsága, a szigorú minőség-ellenőrzés azonban biztosítja, hogy ez a változékonyság nem befolyásolja a készítmény hatásosságát és biztonságosságát.
A felcserélhetőség azt jelenti, hogy egy adott gyógyszerről tetszőlegesen váltani lehet egy másikra, amely várhatóan azonos klinikai hatást biztosít. Eszerint a referencia-gyógyszer felcserélhető a biohasonlóra (vagy fordítva), illetve egyik biohasonló készítményről váltani lehet bármelyik másikra.
A felcserélés módjai:
• gyógyszerváltás, ha a gyógyszert rendelő orvos azonos terápiás célra az aktuálisan alkalmazott gyógyszer helyett egy másikat ír fel.
• helyettesítés (automatikus!), ha az expediáló gyógyszerész a vényen szereplő gyógyszerrel ekvivalens, azzal felcserélhető gyógyszert ad ki a betegnek, a gyógyszert rendelő orvossal való konzultáció nélkül. Magyarországon sem generikus, sem biohasonló készítmények esetében nem működik, néhány kivételtől eltekintve.
Az EU nem szabályozza a referenciakészítmény és a biohasonló gyógyszer felcserélhetőségét, az ilyen készítmények közötti terápiaváltás és az egymással való helyettesíthetőség tagállami hatáskörbe tartozik. (https://ogyei.gov.hu/dynamic/biohasonlo_allasfoglalas_20180212_Final_jav1_20180301.pdf)
A biológiai gyógyszerek esetén kötelezően vizsgálni kell az immunogenitást. Ennek oka a fehérjék és az egyéb biológiai gyógyszerek azon jellemző sajátsága, hogy nem kívánt immunreakciót válthatnak ki, ami egyes esetekben súlyos nemkívánatos hatásokat (pl. anaphylaxiás reakciót vagy késői típusú túlérzékenységi reakciót) idézhet elő vagy a klinikai hatás csökkenéséhez vezethet.
A biohasonló gyógyszer akkor is engedélyezhető a referencia- gyógyszer jóváhagyott javallataiban, ha a biohasonló készítmény klinikai vizsgálatából származó adatok nem állnak rendelkezésre (a javallatok extrapolálása). Erre akkor van lehetőség, ha az összehasonlító vizsgálatokból származó összes tudományos bizonyíték igazolja a biohasonlóságot, és az extrapolált indikációkhoz kapcsolódó specifikus kérdésekre is megnyugtató választ ad (pl. hatásmechanizmus, az adott javallatra potenciálisan specifikus biztonsági és immunogenitási jellemzők).
Az adatok más indikációs területekre történő extrapolálását minden esetben jó minőségű fizikai-kémiai vizsgálatokkal, illetve az összes lehetséges hatásmechanizmus értékelésére alkalmas in vitro vizsgálati adatokkal kell alátámasztani.
(Forrás: OGYÉI)
GY.I.K.
Gyakori kérdések
Mi jelent pontosan a biohasonló gyógyszer?
(Forrás: OGYÉI)
A biohasonló gyógyszer fogalma olyan gyógyszert jelöl, amely nagymértékben hasonló valamely, az EU ban már forgalomba hozatali engedéllyel rendelkező biológiai gyógyszerhez (az ún. referenciakészítményhez). Engedélyezett biohasonló gyógyszert a referenciakészítmény szabadalmi védettségének lejárta után (általában 10 év) lehet forgalomba hozni.
Mivel a biohasonló gyógyszer a biológiai gyógyszerek egyik típusa, ezekre is érvényesek mindazok az előírások, amelyek a biológiai készítményekre vonatkoznak.
Az előállításukhoz használt biológiai forrás természetes változékonysága és a gyártóspecifikus előállítási eljárás különbségei miatt lehetnek kisebb eltérések a biohasonló és a referenciagyógyszer között. A gyártás során szigorú minőség-ellenőrzéssel garantálják, hogy a variabilitás sem a gyógyszer hatását, sem a biztonságosságot nem befolyásolja. Az eltérések tehát sem a biztonságosság, sem a hatásosság tekintetében nem minősülnek klinikailag jelentősnek.
(https://ogyei.gov.hu/dynamic/biohasonlo_gyogyszerek_EMA.pdf)
Van-e különbség a biohasonló- és a biológiai (referencia) gyógyszerek között?
(Forrás: OGYÉI)
Mivel a biohasonló gyógyszereket élő organizmusok felhasználásával állítják elő, kismértékben eltérhetnek a referenciakészítménytől. Ezek a kisebb eltérések azonban klinikai szempontból nem jelentősek, vagyis sem a biztonságosság, sem a hatásosság terén nem várhatók eltérések az alkalmazásuk során. A természetes variabilitás a biológiai gyógyszerek jellemző sajátsága, a szigorú minőség-ellenőrzés azonban biztosítja, hogy ez a változékonyság nem befolyásolja a készítmény hatásosságát és biztonságosságát.
A biohasonló gyógyszerek tényleg olcsóbbak mint az eredeti termékek?
A biohasonló készítmények előállítása sokkal komplexebb folyamat, ezért az áruk sem tér el olyan mértékben mint a klasszikus generikus termékek ára az originális készítményhez viszonyítva.
Ahogy számos EU országban, úgy Magyarországon is speciális szabályozás vonatkozik a biohasonló készítmények árazását tekintve. Az első támogatott biohasonló készítmény az eredeti biológiai készítményhez képest 30%-al alacsonyabb áron tud piacra lépni, utána – 10% és -10% árelőnnyel a soron következő biohasonló.
Hogyan történik a biohasonló gyógyszer regisztrációja az EU-ban?
Az Európai Unióban a gyógyszer-engedélyezés szigorú jogi keretrendszerben történik, amelyen belül 2004-ben lépett érvénybe a biohasonló gyógyszerek engedélyezésére vonatkozó szabályozás. Az EU élen jár a biohasonló gyógyszerekre érvényes jogszabályrendszer lefektetésében azóta, hogy 2006- ban forgalomba hozatali engedélyt kapott az első ilyen típusú gyógyszerkészítmény (egy növekedési hormon, a somatropin készítménye).
A biotechnológiai úton előállított gyógyszerek, illetve meghatározott terápiás területek (pl. daganatos, neurodegeneratív és autoimmun betegségek) gyógyszerei kizárólag az EMA általi centrális eljárás keretében kaphatnak forgalomba hozatali engedélyt az Európai Unióban. Az EU-ban engedélyezett biohasonló gyógyszerek csaknem mindegyikét centrális eljárás keretében hagyták jóvá, mivel ezek előállítása biotechnológiai eljárásokkal történik. Van ugyanakkor néhány biohasonló gyógyszer, amely akár nemzeti eljárás keretében is engedélyezhető – ilyenek például egyes LMWH-készítmények, amelyek sertés bélnyálkahártyájából kivont kis molekulatömegű heparint tartalmaznak.
Ha egy cég az EMA-hoz nyújt be forgalomba hozatali engedély iránti kérelmet, a dokumentációt az EMA humán gyógyszerekkel, illetve gyógyszerbiztonsággal foglalkozó tudományos bizottságai (CHMP, PRAC), valamint a biológiai gyógyszerekkel foglalkozó uniós szakértők (Biológiai gyógyszerekkel foglalkozó munkacsoport, angolul: Biologics Working Party) és a biohasonló gyógyszerekkel foglalkozó szakértői testület (Biohasonló gyógyszerekkel foglalkozó munkacsoport, angolul: Biosimilar Working Party) értékelik.
Az EMA által végzett szakértői értékelés eredményeként megszülető tudományos vélemény az Európai Bizottság elé kerül, amely végül kiadja az EU összes tagországára érvényes forgalomba hozatali engedélyt. (https://ogyei.gov.hu/dynamic/biohasonlo_gyogyszerek_EMA.pdf)
Mikor juthat egy uniós beteg biohasonló gyógyszerhez?
(Forrás: IQVIA)
Amint a biológiai készítmény szabadalma lejár és a nemzeti hatóságok engedélyét is megkapta, továbbá a finanszírozási feltételek is adottak a betegek hozzáférése érdekében.